| |

Protein Folding Using Machine Learning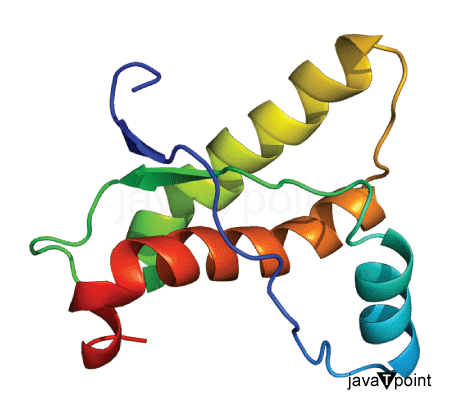
Proteins are like superheroes in our body, playing crucial roles in supporting the functions of our tissues, organs, and overall body processes. These incredible molecules are composed of 20 different building blocks, each one known as an amino acid. It's mind-blowing to think that within our body, there exists a vast array of proteins, each possessing a unique sequence of dozens or even hundreds of amino acids. The fascinating part is that the specific sequence of amino acids in a protein is like a secret code that determines its superpowers, such as its functions. This sequence actually dictates the protein's 3D structure and how it behaves under different circumstances. And guess what? This unique 3D structure then defines the protein's special role in various biological processes. So, it's not just any ordinary code; it's like a super blueprint that shapes the protein's form and unleashes its extraordinary functions, making it an essential aspect of how our body works. But that's not all! Let's delve into the captivating world of protein folding. Picture this: proteins are like masterpieces, formed by long chains of amino acids, and their 3D structure holds the key to unlocking their powers. The process of protein folding is like an intricate dance, where the protein chain elegantly and precisely folds into its own extraordinary and functional shape. It's like the protein discovers its true identity, revealing its unique and powerful abilities to fulfill its mission in our body. Understanding protein folding is no easy feat, though. It's a complex puzzle to solve, given the immense intricacies of the process and the countless possible ways a protein can adopt its shape. But scientists are on a quest to unlock this mystery, as it holds the key to predicting protein structure, which has far-reaching implications in fields like discovering new medicines, researching diseases, and even advancing bioengineering. Understanding it is essential because it directly connects to predicting protein structures. This prediction has broad implications in drug discovery, disease research, and bioengineering. However, protein folding poses challenges due to its complex nature and the countless possible conformations that a protein can take. Machine learning algorithms can be trained on existing protein folding data to learn patterns and relationships between protein sequences and their corresponding structures. These algorithms can then be used to predict the structure of new proteins based on their amino acid sequences. By analyzing large datasets of known protein structures, machine-learning models can uncover hidden patterns and principles that govern protein folding. Benefits of Machine Learning in Protein FoldingHere are some of the benefits of machine learning in understanding Protein folding:
Disadvantages of Protein Folding prediciton using Machine LearningWhile protein folding prediction using machine learning offers numerous advantages, there are also some challenges and disadvantages associated with this approach, that are:
Protien Folding Prediction in Machine Learning Using PythonAbout the DatasetThis dataset contains protein information retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB). The PDB archive is a vast collection of data that includes atomic coordinates and other details about proteins and important biological macromolecules. To determine the location of each atom within the molecule, structural biologists use various methods like X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy. Once they obtain this information, they deposit it into the archive, where it is annotated and made publicly available by the wwPDB. The PDB archive is constantly growing as research progresses in laboratories worldwide. This makes it an exciting resource for researchers and educators. It provides structures for many proteins and nucleic acids involved in crucial life processes, including ribosomes, oncogenes, drug targets, and even entire viruses. However, due to the vastness of the database, it can be challenging to navigate and find specific information. There are often multiple structures available for a single molecule or structures that are partial, modified, or different from their natural form. Despite the challenges, the PDB archive remains a valuable source of data for the scientific community, offering a wealth of information about the structures of various biological molecules. Researchers and educators can explore this vast repository to gain insights into the intricacies of proteins and other macromolecules, supporting advancements in the field of structural biology. ContentThere are two data files. Both are arranged on the "structureId" of the protein:
Now, we will try to make a model that can predict protein structure. Code:
Output: 
We utilized sidechainnet for training our machine learning models, aiming to predict protein structure (angles or coordinates) based on the given amino acid sequences. These examples are almost at the minimum level required for comprehensive model training. The code here is set to train on the debug dataset by default. However, you have the freedom to modify the call to "scn.load" and select a different SidechainNet dataset, such as CASP12, for further experimentation and training. Here, we will be working with two simplified recurrent neural networks (RNNs) to predict angle representations of proteins using their corresponding amino acid sequences:
The internal RNN processes the amino acid sequences, generating angle vectors for each amino acid. While other models used only 3 angles, we can predict all 12 angles provided by SidechainNet in our case.
When requesting DataLoaders, you will receive a dictionary that maps split names to their respective DataLoaders. Output: 
When batches are yielded, each DataLoader returns a Batch namedtuple object with the following attributes:
Output: 
Output: 
Output: 
Helper functions are small, reusable pieces of code that assist in performing specific tasks within a larger program or script. These functions are designed to simplify complex operations, improve code readability, and avoid code duplication. By breaking down complex tasks into smaller, manageable units, helper functions make the main code more organized and easier to maintain.
Attention layers are important in deep learning models because they help the model focus on the most relevant parts of the data. They work like human attention, where some things are given more importance than others in the learning process.
Here, we are going to train the model, such as the Secondary Protein Structure matrix as input. Model Inputs
PSSMA PSSM, also known as a Position Specific Scoring Matrix or Position Weight Matrix in the context of DNA, represents a matrix that provides specific scores or probabilities for each position in a sequence. It is like a special code that tells us how likely each letter (amino acid) appears at different positions in a secret message (protein sequence). Scientists create this code by comparing many similar secret messages from different creatures. The PSSM helps them understand which letters are important and which ones can change without affecting the message's meaning. It's like having a secret decoder that helps scientists learn more about the secret messages in proteins and how they work. Since PSSMs and sequences both have 20 different pieces of information, the secondary structure has 8 possibilities, and the information content is a single number for each piece; when we put all these together, we need a total of 49 values to represent them correctly. Output: 
Output: 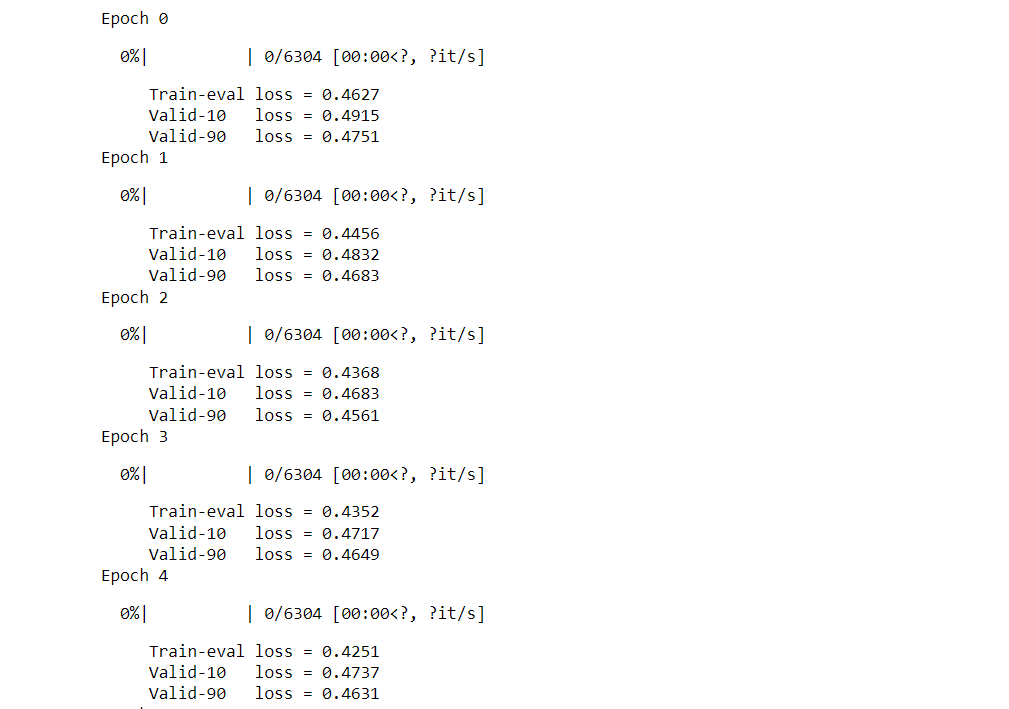
Output: 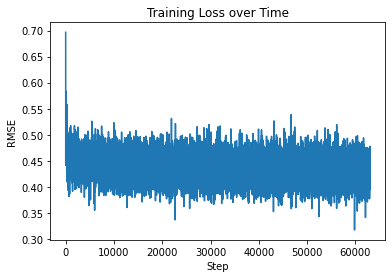
Output: 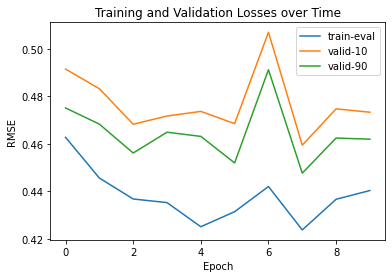
In many situations, we use the scn.BatchedStructureBuilder, which needs two things:
We have a model that knows how to guess the sin and cos values of some angles. But we need the actual angles, not the sin and cos values. So we use a special tool called scn.structure.inverse_trig_transform to change the sin and cos values back into the real angles. Once we have the real angles, we can give them to the BatchedStructureBuilder.
Here, we compare our model's predicted protein structure with the actual protein structure. To make it easier to understand, we visualize these comparisons using 3D plots. Each example has two plots: the top plot shows the model's prediction of the protein structure, and the bottom plot displays the real protein structure. This allows us to see how well our model's predictions match the actual protein structures. Example(01) Output: 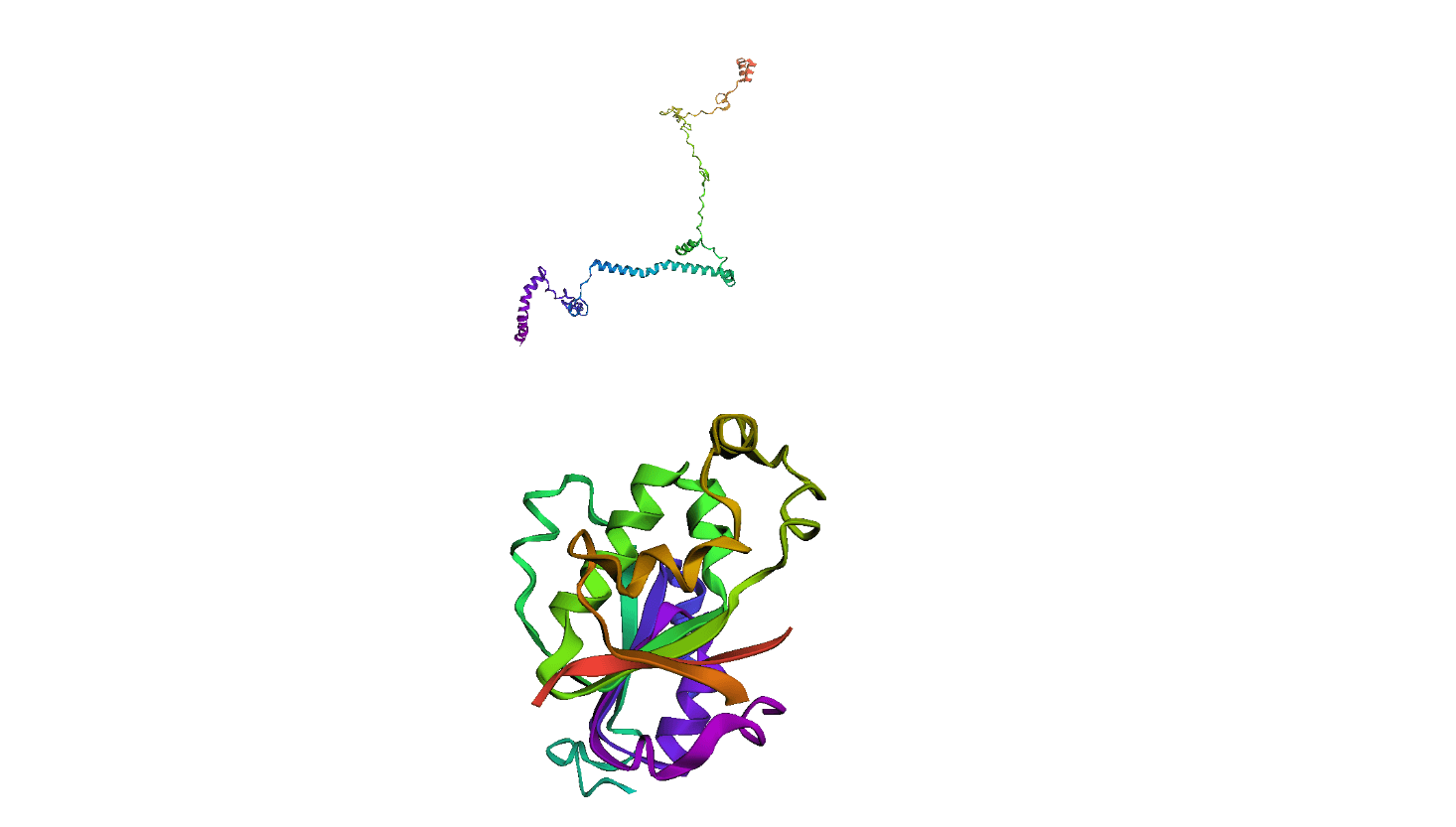
Example(02) Output: 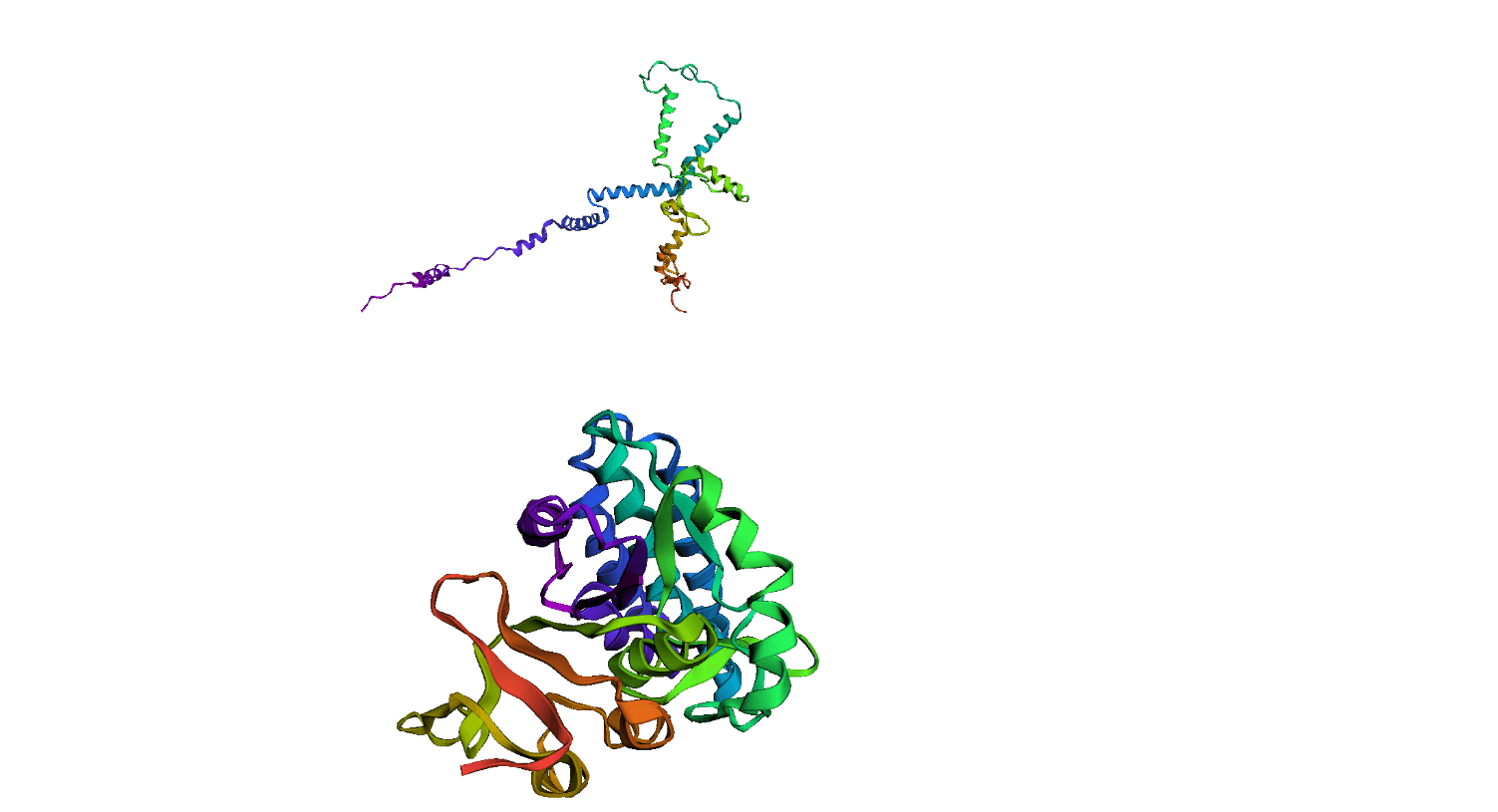
Example(03) Output: 
Example(04) Output: 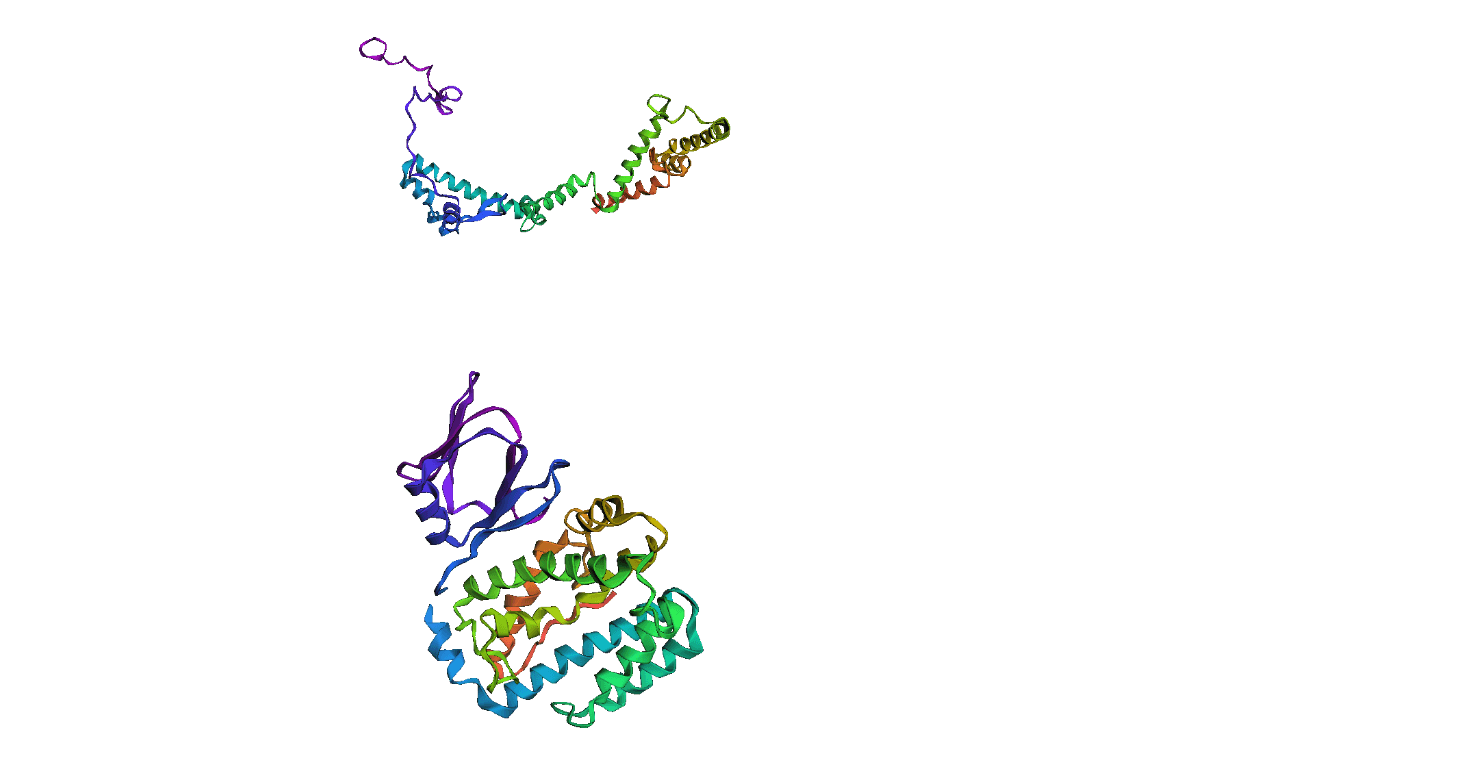
Example(05) Output: 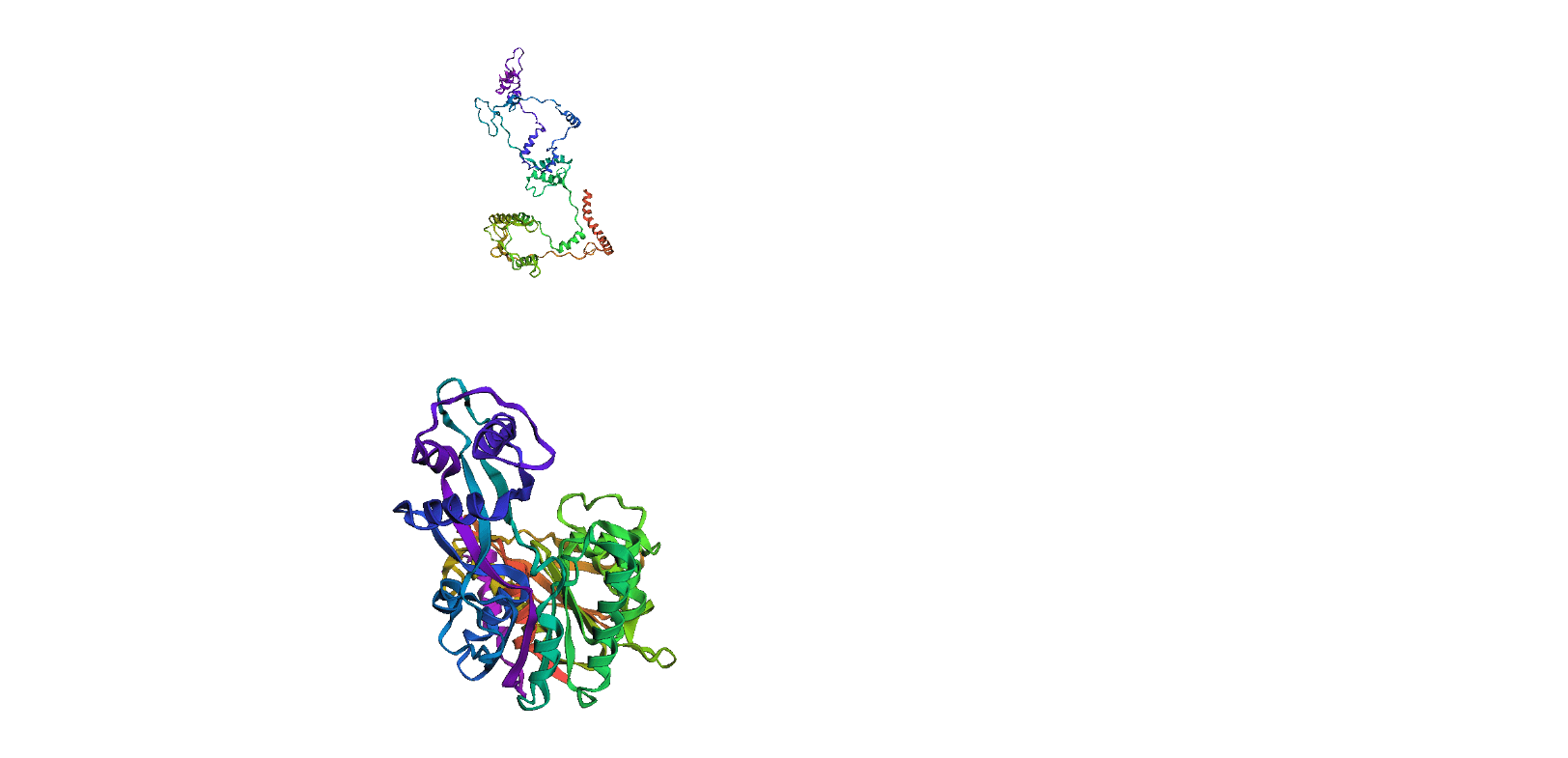
Example(06) Output: 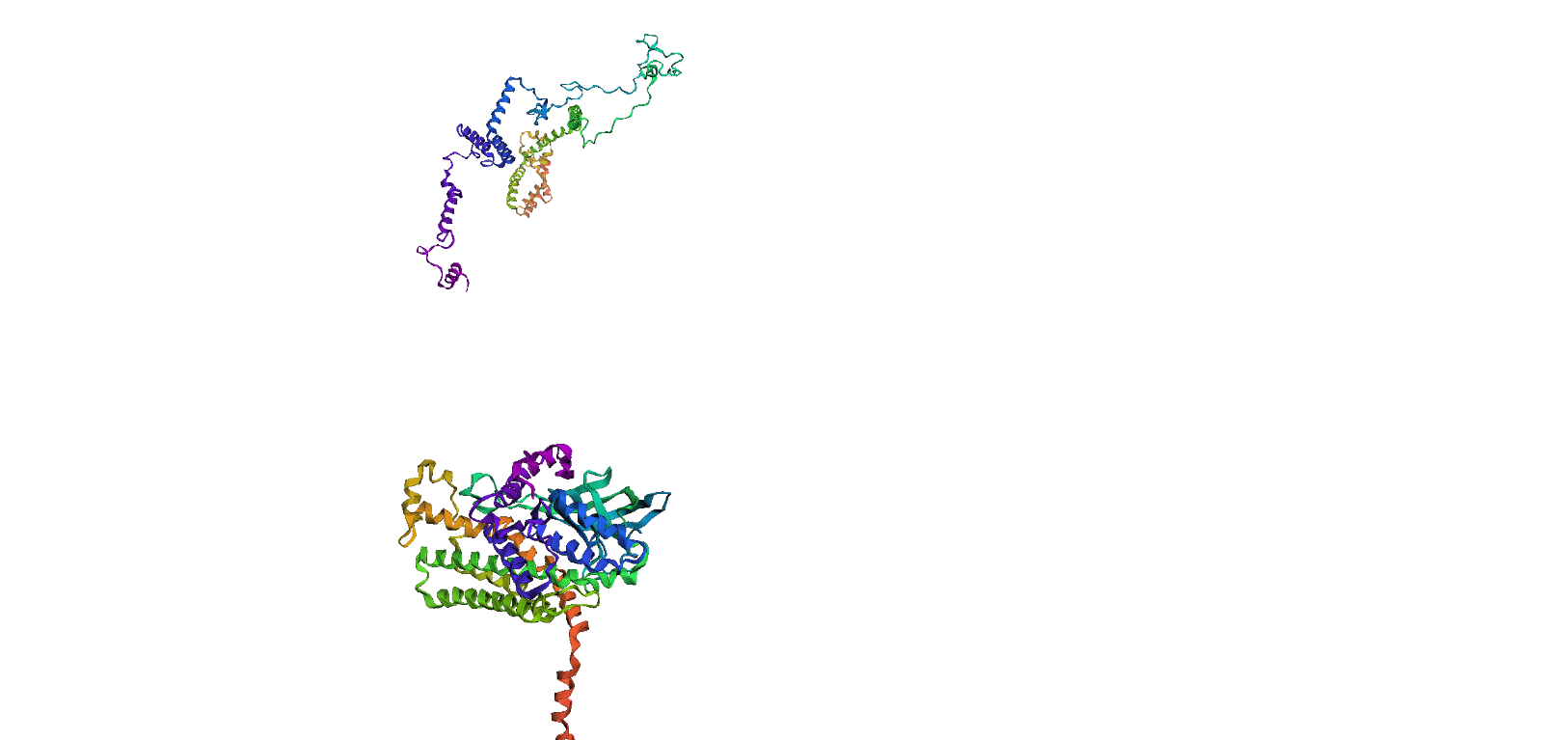
Output: 
Output: 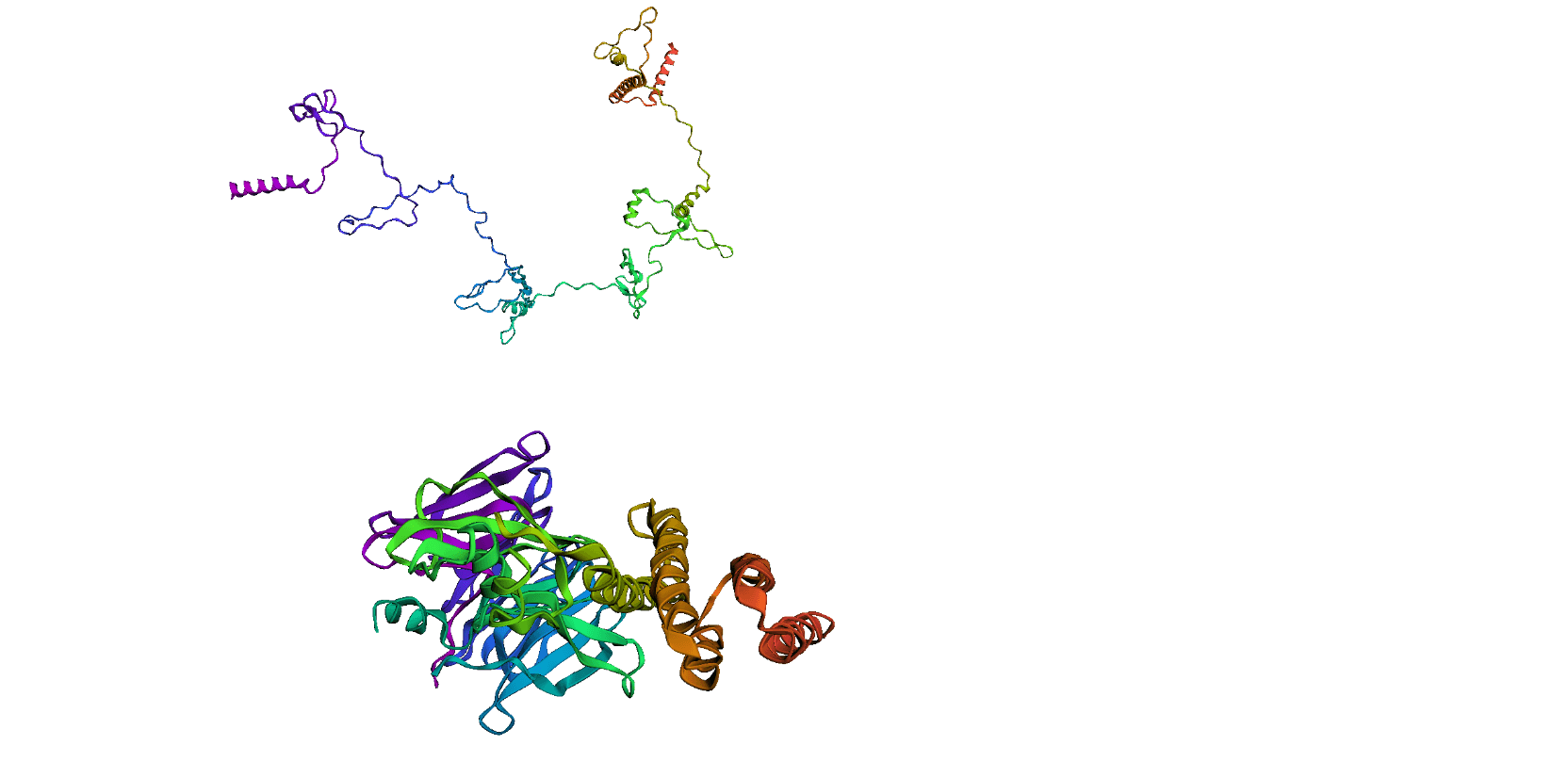
Training(Sequence→ Angles)Now we are going to train the model while taking Protein Sequence as input. Information Flow: Information flow here in a simple Transformer (Attention) model that works with sequence data. The input, represented as [Layers*21], goes through an Embedding layer, resulting in [Layers. Dense Embedding]. Then, it passes through an LSTM layer, transforming into [Layers. Dense Hidden]. Finally, the output comes out of the LSTM and goes through the [Layers Dense Output] layer. Throughout this process, the model processes the input data, extracting relevant information and producing the final output without modifying it. Handling the circular nature of angles: To help our model understand that angles π and -π are the same, we use a special trick. Instead of directly predicting angles, we predict two values for each angle: sin and cos. Then, we use the atan2 function to combine these two values and recover the angles. This way, the model's output will be in the shape of L×12×2, where L is the length of the protein sequence, and the values are between -1 and 1. This approach allows us to handle angles properly and improve the accuracy of our predictions. Output: 

Output: 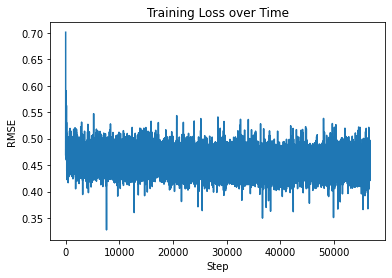
Output: 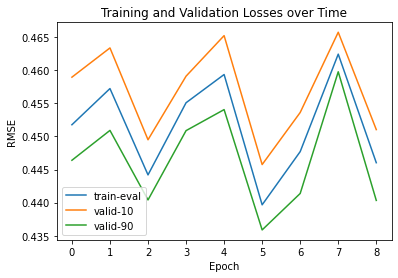
Inference(Sequence-> Angles)Example(09) Output: 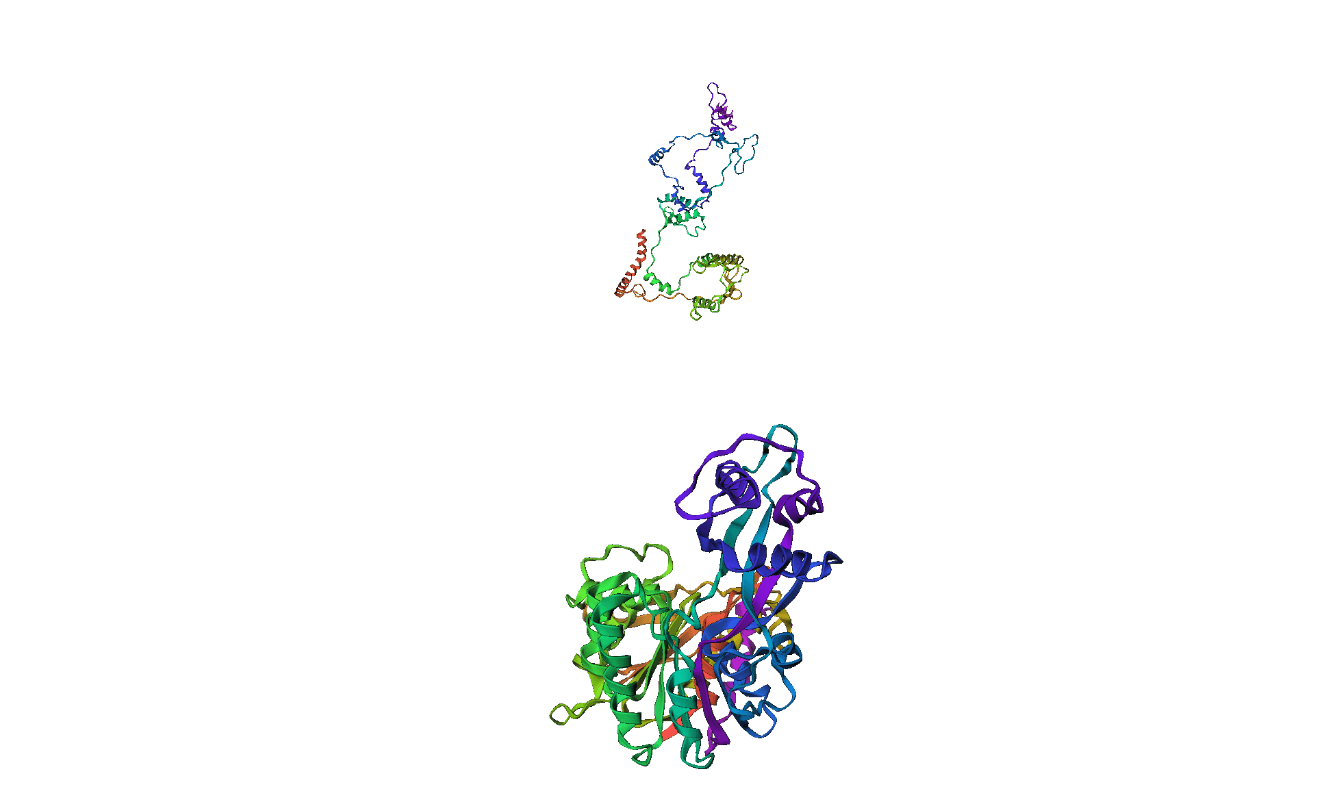
Example(10) Output: 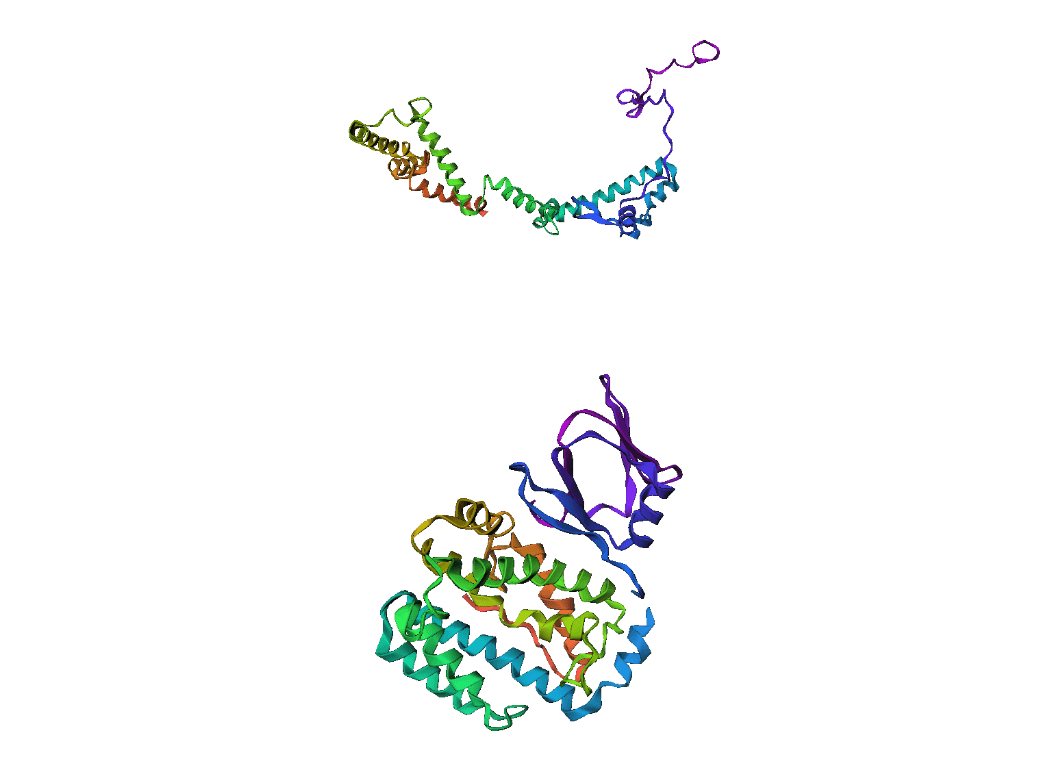
Example(11) Output: 
Example(12) Output: 
Future Aspect of Protein Folding Using Machine LearningThe potential of protein folding in machine learning for the future is incredibly promising, and it has the capability to transform our comprehension of protein structure and function. By harnessing the capabilities of machine learning and adopting interdisciplinary strategies, we stand on the verge of discovering fresh opportunities and expanding the frontiers of scientific exploration. As we persist in uncovering the enigmas surrounding protein folding, we embark on a path of revolutionary research and pioneering applications that will have profound effects on human well-being and beyond. ConclusionProtein folding stands as a critical and intricate process that profoundly impacts the behavior and functions of proteins. The fusion of machine learning with bioinformatics presents an exciting avenue to delve into this intricate world, equipping us with the ability to predict protein structures with unparalleled accuracy. The journey into machine learning and bioinformatics promises to uncover transformative discoveries that will revolutionize medicine and biotechnology. As we venture forth, the enigma of protein folding becomes closer to being unraveled, revealing the profound intricacies of life itself. With machine learning as our ally, we inch ever closer to unveiling the secrets that lie within the realm of protein folding and its vast implications in the grand tapestry of life. |
 For Videos Join Our Youtube Channel: Join Now
For Videos Join Our Youtube Channel: Join Now
Feedback
- Send your Feedback to [email protected]
Help Others, Please Share








